Water Molecular Dynamics Simulation
A downloadable WaterFormationMD for Windows
Overview
“Water Molecular Dynamics Simulation” is a physics-driven application built in C++ using a custom simulation engine. The program utilizes state-of-the-art algorithms to calculate interaction forces, including bond stretching, angle bending, and non-bonded interactions, providing a somewhat accurate representation of water formation at the molecular level. For curiosity, this simulation offers a window into the complex interplay of forces that govern molecular behavior.

Features
- Dynamic Molecular Interactions
- For O–H bonding forces, we use harmonic potentials, where the code calculates bond forces (F = –k_bond · (r – r₀)) to keep bonds near an equilibrium length, simulating the fine balance of molecular forces.
- The simulation models the iconic H–O–H bond angle (approximately 104.5° for water) using a harmonic potential. The Hydrogens should adjust their positions to maintain a natural water-molecule geometry. (Not yet perfect)
- For non-bonded interactions, realistic Lennard-Jones and Coulomb force calculations ensure that even atoms not directly bonded interact in a scientifically sound manner, adding depth to the simulation’s realism.
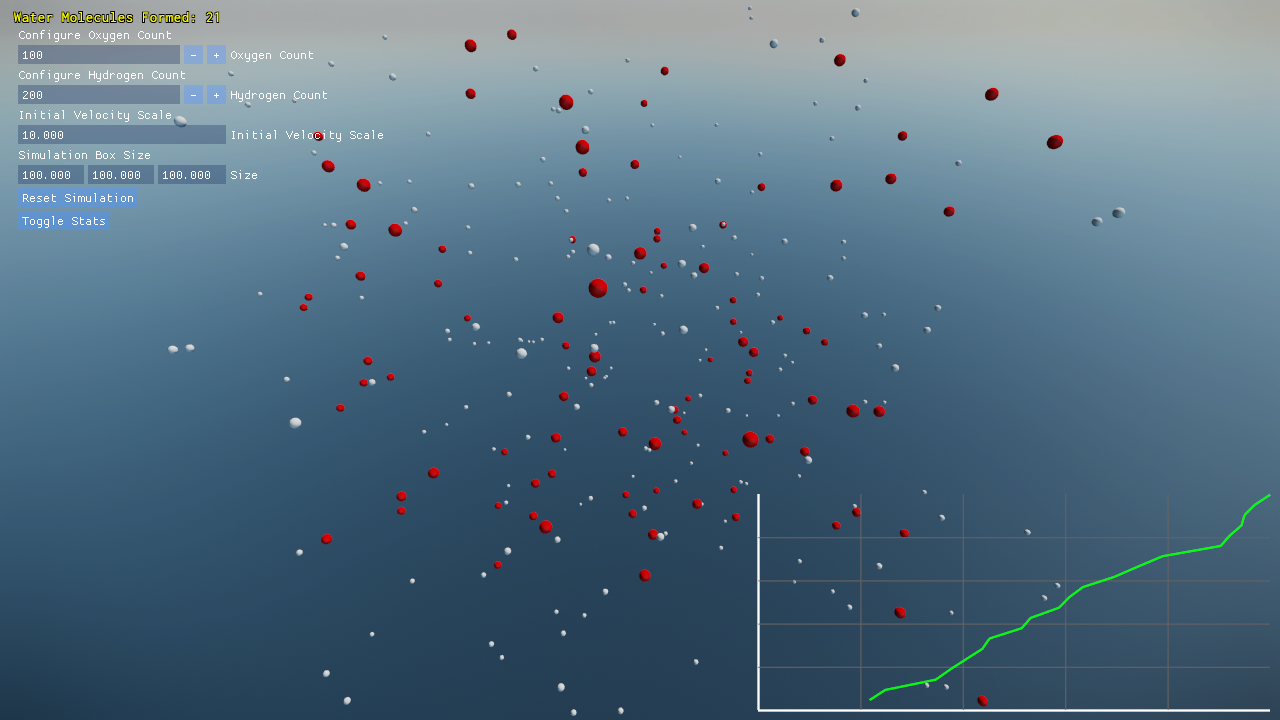
Settings
You may modify key simulation parameters in real-time through an intuitive ImGui interface:
- Configure the number of oxygen atoms (default 100) and hydrogen atoms (default 200).
- Adjust the initial velocity scale to influence the atoms’ kinetic energy.
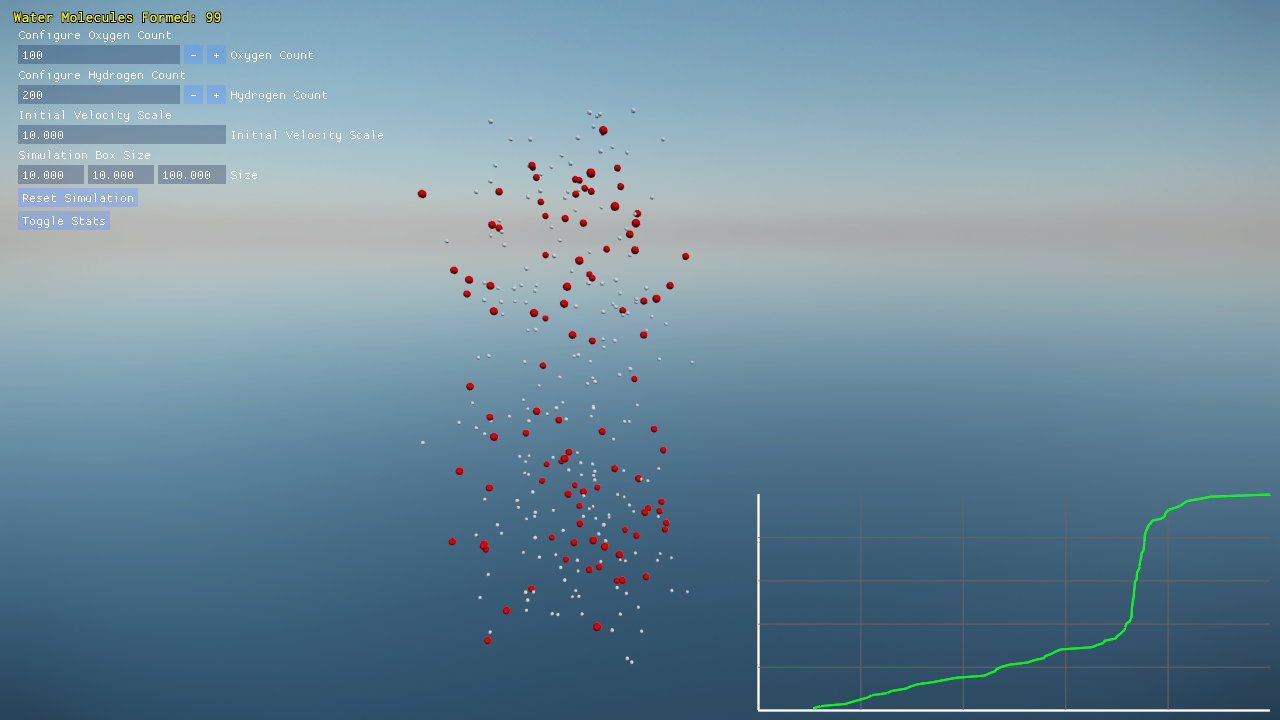
- Dynamically resize the simulation box to observe changes in molecular density.
- Reset the simulation and watch as the scene comes alive with freshly spawned atoms and newly formed bonds.
Visualization and Debugging
- A built-in graph plots the progress of water molecule formation over time, transforming raw data into a visual narrative of the simulation’s evolution.
- Real-time debugging and performance data (such as frame rate) are overlaid, giving you insight into both the simulation’s dynamics and its underlying computational efficiency.
Controls and Interface
- Camera Navigation, switch viewpoints with smooth transitions, and explore the 3D simulation space using typical WASD and mouse input.
- UI Interaction, the settings panel allows you to:
- Modify atom counts and initial velocities.
- Adjust simulation box dimensions.
- Trigger simulation resets or toggle on-screen stats.
A dynamic graph and debug renderer keep you informed, drawing critical simulation data directly onto the screen.
Shortcut Keys
- F12: Capture a screenshot.
- F2: Open the debug console (use Tab to view options and activate auto-complete).
- F5: Display frames per second (FPS).
How It Works
At its core, the simulation computes forces on each atom based on:
- Bonded Forces:
- Harmonic Bond Stretching, each bond strives to maintain an equilibrium distance as described by the equation V₍bond₎ = ½ · k_bond · (r – r₀)².
- Angle Bending, the unique H–O–H angle is maintained using a harmonic potential that pushes the molecular configuration toward 104.5°. (Work in progress)
- Non-Bonded Forces:
- Lennard-Jones Potentials, emulating van der Waals forces with repulsive and attractive interactions.
- Coulomb Forces, adding realistic electrostatic interactions based on effective charges (e.g., –0.834 for oxygen, 0.417 for hydrogen).
- Dynamic Bonding Criteria
- Atoms form bonds based on proximity or high relative acceleration, mimicking energetic collisions that trigger molecular formation. Once an oxygen atom bonds with two hydrogens, the simulation even initializes subtle orbital motions to represent a stable water molecule.
Technical Details
The simulation is written in C++ and leverages a multi-threaded custom engine that integrates modules for rendering, physics, input management, and resource handling.
Although focused on water, the core architecture is modular enough to simulate other molecular systems. The current placeholder for torsional forces hints at future expansion possibilities where more rigorous derivations replace simplified models.
Download the simulation now and step into the realm of molecular dynamics.
Platforms and System Requirements
A modern desktop system with support for DirectX 11 is required to fully experience the detailed 3D rendering and interactive simulation.
Primarily for Windows, with possibilities for cross-platform support with minor modifications (we have Vu.
Future Updates
- Future iterations might include more accurate torsional dynamics or extended non-bonded interactions.
- Exploring simulations for organic molecules or even complex multi-atom reactions.
Share your ideas, tweak parameters, and collaborate to push the simulation’s boundaries further. The community hub and discussion boards are open for all who want to geek out over molecular dynamics!
Download
Install instructions
The download and installation process has been simplified.
- Download the ZIP file.
- Go to the Itch.io page and click the "Download ZIP" button.
- Unzip the File.
- Once downloaded, locate the ZIP file.
- Right-click and select "Extract All" (on Windows) or double-click the file to extract its contents.
- Run the Simulation.
- Open the extracted folder.
- Locate and double-click the executable batch file (e.g., `WaterSimulation.exe`) to launch the simulation.
Leave a comment
Log in with itch.io to leave a comment.